Help
Contents
- General
- Entering the query data
- Setting Bagheera's gene prediction options
- Viewing the results of gene prediction
- Viewing the results of tRNA prediction
- Predicting the most probable decoding scheme
- Bagheera's reference data
| General | Back to top |
This webserver is designed to detect whether a species uses the yeast alternative codon usage or not. For the codon usage estimation, you need to upload genome data. In this data, cytoskeletal and motor proteins are predicted and aligned based on reference data from CymoBase. The aligned predicted CUG positions are the basis for the codon usage prediction.
| Entering the query data | Back to top |
Use the file upload button to upload your query data. Query data need to be complete genome files or unique transcriptome clusters, and must be in fasta format. Please note that the file upload is restricted to 25MB file size. To upload larger files, please contact us.
| Setting Bagheera's gene prediction options | Back to top |
Options for gene prediction
After you have entered the query data, you can set options for gene prediction.The query data are transformed into a BLAST database. TBLASTN searches are preformed for one representative of each reference protein. Representative genes are chosen based on the species selected as an feature model for AUGUSTUS. The genomic regions of the BLAST hits are extended by 500 nucleotides in both directions to obtain better and more complete ab initio gene predictions. To use BLAST with a low complexity filter, check this option.Bagheera predicts cytoskeletal and motor protein genes in the query data with a standalone version of AUGUSTUS. Select the species feature model most suitable to predict genes in your query data from the menu. Protein profiles generated from the reference proteins are used to enhance the quality of the gene predictions.
Options for alignment generation
Bagheera aligns the predicted genes to the respective protein families and classes. This is crucial for the mapping of CUG codons in the predicted genes. You can choose between different alignment methods. MAFFT will add the predicted sequence to the reference multiple sequence alignment, while all other algorithms will perform pairwise alignments between the predicted sequence and a reference sequence.This sequences is selected based on the species which was specified as AUGUSTUS species model. In a second step, the aligned predicted sequence is mapped onto the reference multiple sequence alignment. Pairwise alignment methods are used as implemented in the SeqAn library.
Options for tRNA scan
Bagheera performs a tRNA scan and compares it with reference data containing Ser-tRNACAG and Leu-tRNACAG. tRNAscan-SE is used with a general model or a model specific for eukaryotes to predict the tRNACAG. A BLAST search is performed to find the most similar reference tRNAs. BLAST search will be preformed using a low complexity filter, if this option was selected. In addition, the predicted tRNACAG is aligned with reference tRNAs. To obtain an alignment covering both Ser-tRNACAG as well as Leu-tRNACAG, the alignment will be performed using MAFFT, regardless the specified alignment method for gene prediction.
| Viewing the results of gene prediction | Back to top |
In the results section, you can view a short summary about the predicted CUG codons and the suggested codon usage. Beneath, the detailed results for every predicted cytoskeletal and motor protein are listed. You can toggle the display of the predicted protein itself with all CUG positions highlighted as well as the alignment between predicted protein and reference proteins. All CUG codons in the predicted sequence are translated as leucine, regardless the suggested codon usage.
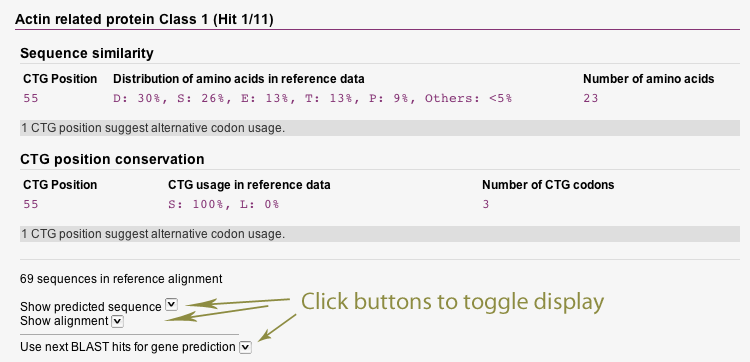
Additionally, information about reference data is shown for every predicted CUG codon mapped to the reference data. This information cover both the distribution of amino acids in reference data as well as the encoding of CUG codons conserved between reference proteins and the predicted protein. The colouring of lines indicates the most probable translation scheme.
The gene prediction is based on the first hit of a TBLASTN search of a reference protein in the uploaded query data. You can restart the gene prediction for a specific protein family by clicking the “Use next BLAST hit for gene prediction” button. The next ten BLAST hits will then be used for gene prediction.
| Viewing the results of tRNA prediction | Back to top |
tRNACAG sequence and secondary structure prediction is displayed. In the secondary structure prediction, unmatched nucleotides are displayed as ".", while a pair of matched nucleotides is indicated by "<" and ">". In addition, tRNAscan score is given. The most probable identity can be assessed based on the BLAST best hits and the alignment with reference Ser-Leu tRNAs. Results of the BLAST search as shown as table indicating the best hits, their bit score and E-value and as BLAST alignment. Table rows are coloured according to the most probable tRNA identity.
| Predicting the most probable decoding scheme | Back to top |
The prediction of the most probable decoding scheme is based on the CUG codon usage and amino acid distribution at the respective alignment position in the reference data.
On the one hand, the amino acid distribution of the reference data is analysed. This is done only for those columns of the multiple sequence alignment, which are aligned with CUG codons in the predicted genes. The distribution of all amino acids accounting for more than 5% of the reference data is listed in the view. Prediction of the codon usage is done based on the chemical properties of the amino acids. The hydrophobic amino acids valine, isoleucine, leucine, methionine and phenylalanine suggest the translation of the predicted CUG as leucine (standard nuclear code). The presence of the polar and small amino acids serine, threonine, cystein and alanine on the other hand suggest the translation of the predicted CUG as serine. However, only portions of these data are taken as hint for one or the other codon usage. A predicted CUG position is considered as discriminative if either hydrophobic or polar amino acids account for the majority of all amino acids. Discriminative positions are coloured in black, while non-discriminative positions are coloured in grey. Discriminative CUG positions are coloured in purple if they alternative codon usage and orange if the suggest standard codon usage. A predicted CUG position is considered as non-discriminative if neither hydrophobic nor polar amino acids account for the majority of all amino acids. It is then displayed in grey.
On the other hand, CUG codons conserved between the predicted protein and the reference data are taken as hint. The translation of CUG as serine in the reference data suggests the translation of the predicted CUG as serine as well. Likewise, the translation of CUG as leucine in reference data suggests the translation of the predicted CUG as leucine as well.
The number of CUG positions suggesting alternative codon usage or standard codon usage is indicated for each predicted protein separately. In addition, the overall number is listed in the summary section.
| Bagheera's reference data | Back to top |
The reference data are manually annotated and curated cytoskeletal and motor proteins of the Saccharomycetes clade. We have annotated more than 2300 sequences from 26 protein families. Out of these protein families, actin-related proteins, capping proteins, kinesins, myosin heavy chain proteins and tubulins are split up into different classes/subfamilies, leading to a total count of 44 protein families/classes used as separate entities for codon usage prediction. Each of these protein families or classes encodes dozens up to a few hundred CUG codons. We have annotated these sequences in about 80 different species or strains of the Saccharomycetes clade. The reference data are updated from CymoBase once a month.








